

|
医疗器械警戒快讯 2025年第6期(总第220期)
发布日期:2025-07-02
医疗器械警戒快讯
(总第220期)
内容提要
美国FDA发布关于ZOLL Circulation公司因影响心肺复苏术效果的故障召回AutoPulse NXT复苏系统的警示信息 发布日期:2025年6月23日 召回级别:FDA已经确认此次召回为最严重的等级,如果继续使用此产品,可能会导致严重伤害或死亡。此次召回涉及从使用单位或销售机构撤回相关产品。 受影响的产品 产品用途 AutoPulse复苏系统(型号200)是一种自动化、便携式、由电池供电的设备,用于辅助手动心肺复苏术,适用于经历临床死亡(定义为缺乏自主呼吸和脉搏)的成年患者,只有当胸外按压可能对患者有益时,才应使用该产品。 召回原因 ZOLL Circulation公司因故障代码(FC1060)召回AutoPulse NXT复苏系统,该故障代码表明在正确测定按压深度时出现错误,导致按压可能停止或者按压可能不够深,进而影响心肺复苏术(CPR)挽救生命的效果。 使用受影响的产品可能会导致严重的不良健康后果,包括重要器官血流减少、缺氧性脑损伤和死亡。 目前,还没有与此问题相关的伤亡报告。 采取措施: 2025年3月4日,ZOLL Circulation公司向所有受影响的客户发送了紧急医疗器械召回通知,建议采取以下措施: (美国FDA网站)
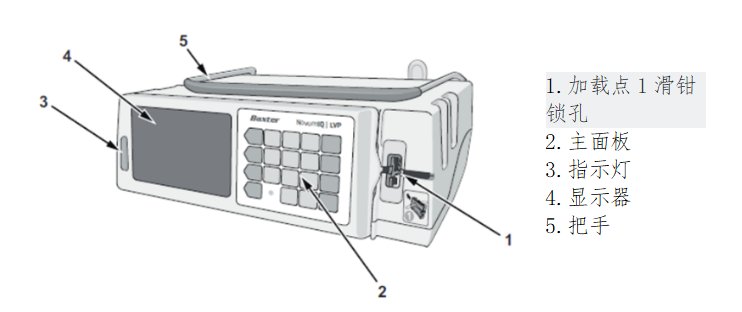
美国FDA发布Baxter公司因潜在的输液不足风险更新Novum IQ大容量输液泵使用说明书的警示信息 发布日期:2025年6月6日 召回级别:FDA已经确认此次召回为最严重的等级,如果继续使用此产品,可能会导致严重伤害或死亡。此次召回涉及使用说明书的更新,不涉及从使用单位或销售机构撤回相关产品。 受影响的产品
产品用途 Baxter Novum IQ输液器泵用于在医生或其他经过认证的医疗保健专业人员的指导或监督下,为患者提供肠外液体、血液和血液制品等静脉注射。 更新使用说明的原因 Baxter公司发现,在开启“待机模式”或者在设备已加载的情况下关闭,Novum IQ大容量泵可能会出现输液不足的情况。将给药装置长时间加载在泵上可能由于该装置的压缩而导致后续输液不足。长时间处于待机模式或关机后,以较高流速注入的风险会增加。 测试表明,当流量大于50 mL/h时,某些输液在2小时30分钟后可能会经历超过10%的流量变化。在最大流速为1200 mL/h、最大待机时间为12小时的最坏情况下,可观察到50%的欠注现象。这可能导致输液不足,包括药物、静脉营养品、血液和血液制品。 需要注意的是,即使有10%的流量变化,儿科患者(婴儿29天至2岁)也可能面临脱水、药物治疗和营养不足以及输血不足等可能导致发病率和死亡率增加的风险。 Baxter公司报告了1例严重伤害报告,但没有与此问题相关的死亡报告。 采取措施 4月24日,Baxter公司向所有受影响的客户发送信函,建议采取以下措施: (美国FDA网站)
美国FDA发布关于Medtronic公司因固定法兰断开风险召回Shiley成人柔性气管造口术导管的警示信息 发布日期:2025年6月5日 召回级别:FDA已经确认此次召回为最严重的等级,如果继续使用此产品,可能会导致严重伤害或死亡。此次召回涉及从使用单位或销售机构撤回相关产品。 受影响的产品 产品用途 Shiley成人柔性气管造口术导管(带TaperGuard套囊可重复使用的内套管),用于通过提供气管通路来帮助患者呼吸。它也可以在称为经皮扩张气管切开术(PDT)的过程中使用,这是一种在颈部形成开口以放置导管的方法。 召回原因 Medtronic公司及其子公司Covidien正在召回Shiley成人柔性气管造口术导管,该导管配有TaperGuard套囊,可重复使用的内套管。由于如果固定法兰断开,导管可能会移位,这可能会阻止患者呼吸和/或阻塞气道,从而导致严重或危及生命的紧急情况。 使用已经从套管固定法兰断开的气管造口术导管可能导致呼吸衰竭、气道组织损伤、窒息(吸入)、呼吸道感染、气道紧缩(支气管痉挛)、延迟治疗和/或死亡。 Medtronic公司尚未报告任何与此问题相关的严重伤害或死亡。 采取措施 在考虑更换时机时,评估患者的总体风险。 继续遵循当前的产品使用说明(IFU)以及工厂特定的策略和程序。 2025年2月26日,Medtronic向所有受影响的客户发送了紧急医疗器械召回通知函,建议采取以下措施: (美国FDA网站)
美国FDA发布关于Zyno Medical公司因软件问题召回部分Z-800系列输液泵的警示信息 发布日期:2025年6月16日 召回类型:FDA已经确认此次召回为最严重的等级,如果继续使用此产品,可能会导致严重伤害或死亡。此次召回涉及从使用单位或销售机构撤回相关产品。 受影响的产品 某些序列号的Z-800系列输液泵,具体见下: 受影响产品的详细情况请参见FDA官方网站。 产品用途 Zyno Medical Z-800输液系统用于在医生或其他经过认证的医疗保健专业人员的指导或监督下,为患者提供肠外液、血液和血液制品的静脉输注。 召回原因 Zyno Medical公司表示,某些发布给客户的Z-800、Z-800F、Z-800W和Z-800WF输液泵软件版本是错误的,这些软件未经过所需验证和确认测试。这些输液泵可能会遇到意想不到的性能问题,包括基本功能和风险控制措施问题,如管道空气检测、流量警报和预防倒流。 管路空气检测不正确、与其他故障相关的错误音频警报等情况带来的最坏情况是可能会导致严重伤害,如药物输送不足或过度、超过1 mL的静脉空气栓塞、未检测到药物或肠外液倒流,以及将“保持静脉开放(KVO)”流速从5 mL/h降低到1 mL/h,这可能导致非常低的KVO流速,并可能导致导管远端形成血凝块,最终导致外周静脉导管(PIV)置管失败。 目前,Zyno Medical公司尚未报告任何死亡或严重伤害。 采取措施 2025年5月7日,Zyno Medical公司向所有受影响的客户发送了一封电子邮件,建议采取以下措施: (美国FDA网站)
美国FDA发布关于Q'Apel Medical公司因抽吸导管远端尖端质量问题召回Hippo 072抽吸系统的警示信息 发布日期:2025年6月17日 召回级别:FDA已经确认此次召回为最严重的等级,如果继续使用此产品,可能会导致严重伤害或死亡。此次召回涉及从使用单位或销售机构撤回相关产品。 受影响的产品:072抽吸系统或Hippo 072抽吸系统,包括Cheetah输送工具和抽吸导管,UDI及批号等参见FDA网站: https://www.fda.gov/medical-devices/medical-device-recalls/aspiration-catheter-recall-qapel-medical-inc-removes-hippo-072-aspiration-system-and-cheetah 产品用途 Hippo 072抽吸系统适用于清除脑部血栓,在脑梗死后8小时内使用。这一产品为无法接受组织型纤溶酶原激活剂静脉注射或对溶栓药物无反应的脑梗死患者提供了一种治疗方法。 召回原因 Q'Apel Medical公司启动此次召回是因为收到了FDA的警告信。FDA在警告信中对抽吸导管在抽吸过程中的远端尖端性能提出了质疑,并对Hippo产品的内部流程和出厂许可范围表示担忧。FDA表示,使用受影响的抽吸导管可能会导致严重的不良健康后果,包括血管痉挛或撕裂,甚至死亡。目前,Q'Apel Medical公司已向FDA报告了两起与此问题相关的伤害事件。目前还没有发现死亡的报告。 采取措施 Q'Apel Medical公司已向所有受影响的客户发送了一封医疗器械召回信,建议立即隔离并退回相关产品。
(美国FDA网站)
|


|