

|
医疗器械警戒快讯 2025年第4期(总第218期)
发布日期:2025-05-15
医疗器械警戒快讯
(总第218期)
内容提要
加拿大Health Canada发布关于GE Healthcare公司因电池问题召回便携式彩色超声诊断系统的警示信息
发布日期:2025年3月26日
召回级别:Ⅱ级
召回产品:便携式彩色超声诊断系统,具体型号和批次见下表:
召回发起时间:2025年3月17日
召回原因:GE Healthcare公司发现某些软件版本为R2、R3和R4的Venue Go和Venue Fit便携式彩色超声诊断系统的电池可能会发生内部故障,从而导致冒烟或火灾。
(加拿大Health Canada网站)
美国FDA发布关于Medtronic公司因公鲁尔头材质脱落风险召回主动脉根部插管的警示信息
发布日期:2025年3月31日
召回级别:FDA已将此次召回确定为最严重的类型如果继续使用该器械,可能会导致严重伤害或死亡。此次召回涉及从医疗器械使用单位或销售机构撤回部分器械。
召回产品:
产品名称:DLP主动脉根部插管;MiAR插管;带通气管的DLP主动脉根部插管
产品UDI/型号及批号详见FDA网站:
https://www.fda.gov/medical-devices/medical-device-recalls/vascular-cannula-recall-medtronic-removes-aortic-root-cannula-due-unexpected-loose-material-male
产品用途:
主动脉根部插管在心肺机(体外循环)手术中使用6小时或更短时间。当旁路手术完成后,套管也可用于从大动脉(主动脉)中取出空气。
召回原因:
Medtronic公司正在召回主动脉根部套管,原因是该套管使用的男性导管存在意外松动物质的风险。松散的材料有可能移位,并给患者带来严重的不良健康后果,包括因延迟治疗、中风和死亡造成的伤害。
目前没有收到关于伤害和死亡的报告。
采取措施:
2025年2月5日,Medtronic向所有受影响的客户发送了一封医疗器械紧急召回信,建议请勿使用受影响批次产品并隔离所有未使用的受影响产品;联系美敦力客服办理退货。
(美国FDA网站)
澳大利亚TGA发布关于Zoll公司因错误代码导致无法提供治疗问题召回G5体外除颤器的警示信息
发布日期:2025年4月10日
召回级别:I级
召回产品:Zoll Powerheart G5半自动和全自动体外除颤器(AED)
召回原因:G5 AED会自动执行自检,以提醒用户可能干扰其提供急救治疗能力的情况。错误代码EC 501是在设备的自动自检过程中生成的特定错误代码,如果显示该代码,则无法提供急救的治疗。
如果用户在尝试使用设备之前没有注意到EC 504或其他设备警报,无法提供急救治疗的设备有给患者造成伤害的潜在风险。
此故障模式(EC501)可能是由于暴露于设备发布的参数之外的湿度环境中造成的。目前调查正在进行中。
召回措施:ZOLL公司正在与相关方传达将有风险的设备存放在适当湿度区域的重要性。客户应该确保设备按照发布的参数要求存储;监控设备自检结果,并采取措施解决任何警报。
(澳大利亚TGA网站)
美国FDA发布关于BD公司血管内PICC导管问题的早期警示信息
发布日期:2025年4月18日
本通讯是加强医疗器械召回计划通讯试点的一部分。FDA已经意识到一个潜在的高风险器械问题。FDA将随时通知公众,并在有重要新信息时更新此网页。
受影响的产品
FDA注意到,BD及其子公司Bard Access Systems已向受影响的客户发出信函,建议使用或销售方移除某些未使用的PowerPICC血管内导管,并且已更新在用的PowerPICC血管内导管的使用说明:
采取措施
2025年3月11日,BD公司向所有受影响的医疗保健提供者发送了紧急医疗器械产品召回通知,建议采取以下行动:
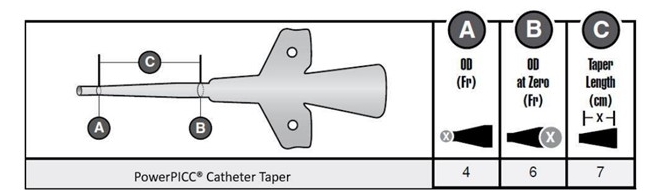
图1:PowerPICC导管锥形区域图像
早期警示的原因
BD公司发现受影响导管的材料疲劳泄漏增加,主要表现为导管体的横向/周向裂纹,见图2。BD公司的调查表明,这是由于用于制造导管的材料树脂的问题。
 图2. 导管体横向/周向裂纹示例
与材料疲劳泄漏相关的风险包括浸润、外渗、不适、静脉炎、出血、空气栓塞、异物栓塞、感染和治疗中断。BD公司报告了10例与此相关的严重伤害。
器械使用情况
PowerPICC导管适用于短期或长期由外周进入中心静脉系统进行静脉治疗,注射造影剂,并监测中心静脉压力。
(美国FDA网站)
美国FDA发布关于Conavi公司诊断性血管内导管问题的早期警示信息
发布日期:2025年3月12日
召回背景
本通讯是加强医疗器械召回计划通讯试点的一部分。FDA已经意识到一个潜在的高风险器械问题。FDA将随时通知公众,并在有重要新信息时更新此网页。
受影响的产品
产品用途
Novasight Hybrid导管是用于冠状动脉血管内成像系统的组成部分。
采取措施
2025年3月12日,Conavi公司向所有受影响的医疗机构发送了紧急医疗器械产品撤回通知,建议采取以下行动:
早期警示原因
根据1起报告事件,Novasight Hybrid导管的鞘在使用过程中脱落并遗留在患者体内。所幸最终通过手术完整取出,未造成进一步伤害。
冠状动脉内脱落的鞘管若未能及时取出,可能导致冠状动脉痉挛、夹层、穿孔、血栓形成、栓塞或血管突然闭塞。
移除过程中可能引发血流动力学后果,如心律传导异常(心动过缓/过速)、低血压、呼吸功能不全。若移除失败,需紧急心脏手术干预。
目前Conavi公司未报告与此问题相关的其他事故或伤害。
(美国FDA网站)
美国FDA发布关于Abbott公司因突发断电问题召回心脏泵配件HeartMate移动电源装置的警示信息
发布日期:2025年4月24日
FDA已将此次召回确定为最严重的类型如果继续使用该器械,可能会导致严重伤害或死亡。此次召回涉及从医疗器械使用单位或销售机构撤回部分器械。
受影响产品
 产品用途
MPU是HeartMate II和HeartMate 3左心室辅助系统的配件,为系统控制器供电,适用于无需监护的家庭/临床环境。该系统通过植入式泵将血液从左心室分流至主动脉。
召回原因
Abbott公司收到MPU突发故障(无法启动/突然关闭并重新启动)报告,系统控制器指示黄色扳手警报或“无外部电源”警报。Abbott公司已确定,此问题与2024年4月至2025年2月期间生产的特定MPU电子元件有关。已出现故障的产品将立即更换,未出现故障的受影响产品将于2025年6月前逐步更换。
若MPU断电,系统控制器备用电池最多维持15分钟泵运转。超时未连接14V电池将导致泵停转,可能引发血流动力学紊乱、血栓或死亡。
目前Abbott公司未收到相关严重伤害或死亡报告。
医疗机构应采取的措施
患者和用户应采取的措施
(美国FDA网站)
 |

|