

|
医疗器械警戒快讯 2025年第3期(总第217期)
发布日期:2025-03-31
医疗器械警戒快讯
(总第217期)
内容提要 FDA发布关于Biosense Webster公司因脑卒中率高或短暂性脑缺血问题召回消融导管的警示信息 美国FDA发布关于飞利浦公司因导致额外手术干预风险召回Tack血管内系统的警示信息 美国FDA发布关于奥林巴斯公司因显影端脱落问题召回一次性使用导管鞘套件的警示信息 美国FDA发布关于Getinge公司因病人和医护人员有暴露于有毒化学物质氟化氢的风险召回蒸发器的警示信息 美国FDA发布关于BD公司因存在加载过时自动化编程请求风险召回Alaris输液泵系统的警示信息
FDA发布关于Biosense Webster公司因脑卒中率高或短暂性脑缺血问题召回消融导管的警示信息
发布日期:2025年2月28日 召回类型:此次召回涉及更新器械的使用说明,不涉及从使用单位或经营企业撤回上述器械。FDA已将此次召回确定为最严重的类型。如果您不按照更新的说明继续使用此器械,可能会导致严重伤害或死亡。 召回产品:
产品名称:一次性使用磁定位心脏脉冲电场消融导管Varipulse Bi-directional Ablation Catheter(Varipulse 平台) 唯一器械标识符(UDI):10846835025460(GTIN) 批次/序列号:所有批次和序列号 产品用途: Varipulse导管适用于基于导管的心脏电生理标测(刺激和记录),当与Trupulse能量平台一起使用时,用于治疗药物难治性、复发性、症状性的阵发性房颤。导管与Carto 3电生理导航系统一起使用时,为其提供位置信息。该器械适用于22岁及以上的成年人。 召回原因: 自2024年11月FDA批准该器械后,首批132例患者手术中出现了高比例脑卒中或短暂性脑缺血发作(TIA),Biosense Webster更新了Varipulse消融导管的使用说明。Biosense Webster将其分销范围限制在少数几家美国医院,以便从医生那里获得更多反馈,这称为“Varipulse美国外部评估”。在美国外部评估期间接受该器械治疗的132名患者中,有4名患者(约3%)在手术后不久出现脑卒中或TIA。这个比例超过了此类手术的预期比例(通常为1%或更低)。 Biosense Webster调查了患者脑卒中或TIA的手术,发现手术中器械的使用方式与产品使用说明中的建议存在一些差异。这些差异可以解释脑卒中或TIA风险较高的原因,包括治疗更严重的心律不齐患者、向心脏输送更多的能量、向心脏的同一位置重复输送能量,以及向临床试验中未研究的位置输送能量。根据现有这些可能将使用差异与脑卒中或TIA联系起来的数据,Biosense Webster在更新后的使用说明中,建议仅按照临床研究的结果使用该器械。 使用受影响的产品可能会导致严重的不良健康后果,包括脑卒中、TIA和死亡。Biosense Webster报告了4起与此问题相关的严重伤害。 召回措施: 在2025年2月14日,强生医疗科技公司(Biosense Webster的母公司)向所有受影响的客户发送了一封紧急医疗器械召回(更正)信,以更新其1月份关于暂停美国外部评估的通信。2月的信中建议医疗保健提供者采取以下行动: 这封信是在2025年1月强生宣布暂时暂停美国外部评估和所有美国Varipulse案件之后发出的。1月的公开信详见以下链接: (美国FDA网站)
美国FDA发布关于飞利浦公司因导致额外手术干预风险召回Tack血管内系统的警示信息
发布日期:2025年3月3日 召回级别:此次召回涉及从医疗器械使用单位或销售机构召回部分器械。美国食品药品监督管理局(FDA)已将此次召回识别为最严重的类型。如果继续使用该器械,可能会导致严重伤害或死亡。 召回产品: 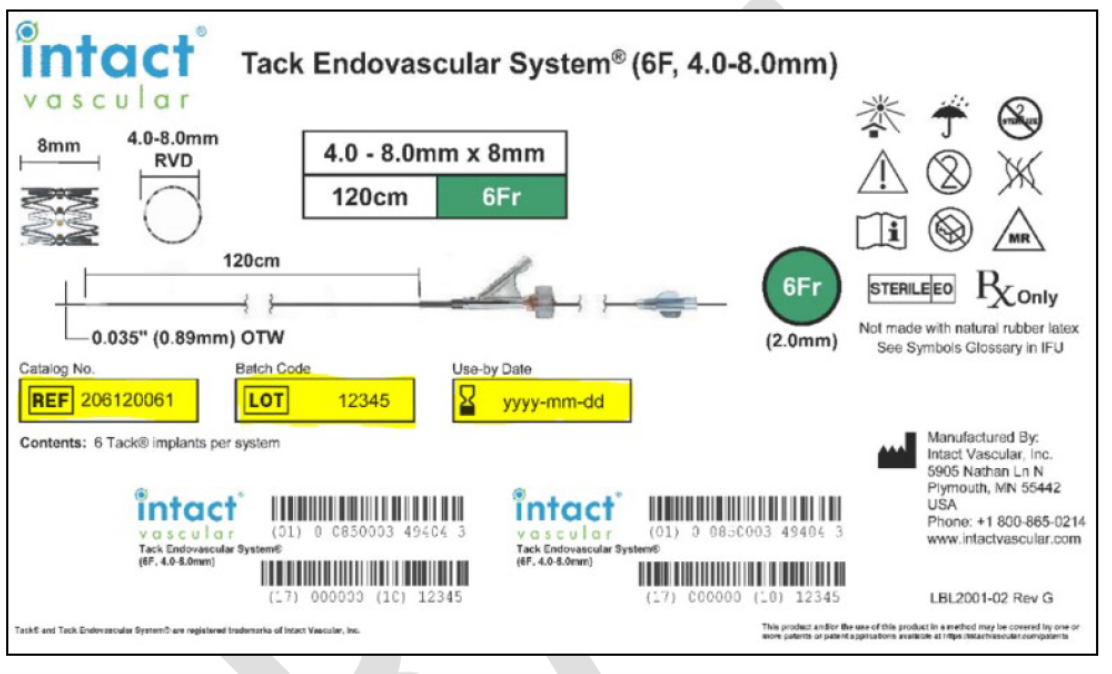 召回原因:飞利浦公司此次召回的Tack血管内系统,用于治疗在外周血管成形术中使用球囊或支架增宽血管后出现的血管夹层,Tack血管内系统可以将受损组织连接到血管内皮以修复夹层。本次召回涉及的Tack血管内系统在使用过程中可能导致严重的不良健康后果,包括血栓、动脉夹层撕裂等短期风险,以及疼痛、组织脱落、再狭窄、需要搭桥手术干预、截肢和死亡等长期风险。目前,已经收到20份伤害报告,还没有收到死亡报告。 召回措施: 2025年1月10日,飞利浦公司向所有受影响的用户发送了一封紧急医疗器械召回信,建议立即停止使用受影响的Tack血管内系统;立即检查库存并隔离所有受影响的器械以防止被使用;在7天内反馈回复表,以便飞利浦公司开始办理退货和退款流程;将本通知转发至与本次召回涉及产品相关的所有用户。飞利浦公司将不再分销涉及产品。 (美国FDA网站)
美国FDA发布关于奥林巴斯公司因显影端脱落问题召回一次性使用导管鞘套件的警示信息
发布日期:2025年2月11日 召回级别:I级 召回产品:一次性使用导管鞘套件,具体型号、UDI参见FDA网站。 产品用途:该产品与奥林巴斯内窥镜配合使用,用于在呼吸器官中采集细胞或组织样本。 召回原因:导管鞘显影组件可能分离并脱落至患者体内。 召回措施:奥林巴斯公司于2025年1月15日通过信函通知了收货方。要求收货方检查库存并隔离受影响的设备、立即停止使用该产品并通过奥林巴斯网络门户确认收到该信函。若产品已进一步分销,需将通知转发给所有相关人员和客户。 (美国FDA网站)
美国FDA发布关于Getinge公司因病人和医护人员有暴露于有毒化学物质氟化氢的风险召回蒸发器的警示信息
发布日期:2025年3月3日 召回级别:FDA已经确认此次召回为最严重的等级,如果继续使用相关产品而不进行纠正,可能会导致严重伤害或死亡。此次召回涉及从使用单位或销售机构撤回相关产品。 这是2024年发布的相关召回信息的补充信息。 召回产品: 产品名称: 七氟醚Maquet填充蒸发器,七氟醚Quick-Fil填充蒸发器 UDI/产品编号: Maquet填充:07325710000212/6682282 Quick-Fil填充:07325710001141/6682285 序列号: Maquet填充:17336–23784和所有大于1339的序列号 Quick-Fil填充:所有大于3761的序列号 产品用途: 七氟醚Maquet填充和七氟醚Quick-Fil蒸发器是流动麻醉系统的一部分。这些蒸发器专门用于容纳、蒸发和混合液体七氟醚和氧气,以开始和维持全身麻醉。制造商Abbvie、Baxter和Piramal公司生产不同配方的七氟醚。 召回原因: Getinge公司在收到与Piramal或Baxter公司生产的低含水量七氟醚一起使用后蒸发器内变色和/或腐蚀的报告后,正在召回七氟醚Maquet填充蒸发器和七氟醚Quick-Fil蒸发器。蒸发器中使用的七氟醚可能降解为氟化氢。如果吸入或接触皮肤,这种有毒有害的酸可能会对患者和医护人员造成危险。这是对之前召回信息的补充。 使用受影响的产品可能会导致严重的不良健康后果,包括呼吸道刺激,导致肺部积液(肺水肿)和/或血液中钙含量严重偏低(低钙血症)、起泡、皮肤伤口(浅表溃疡)、血液中镁含量偏低(低镁血症)和死亡。 目前公司未收到伤害和死亡的报告。 召回措施: 如果任何受影响的蒸发器曾经可能与Piramal或Baxter公司生产的七氟醚一起使用过,请不要使用。 不要将七氟醚留在任何Getinge公司的蒸发器中超过30天,在外部运输过程中蒸发器中也不要留存七氟醚。 确保任何含有七氟醚的Getinge蒸发器都是在过去30天内使用的。 不要清空任何有变色或腐蚀迹象或有异常气味的蒸发器。 如果含有七氟醚的Getinge蒸发器在过去30天内没有使用过并且没有腐蚀或变色的迹象,则清空并干燥运行(见下文)。 2025年1月15日,Getinge公司及其子公司Maquet Critical Care AB公司向所有受影响的客户发送了一封紧急医疗器械召回函,建议采取以下措施: 1.隔离所有受影响的产品。 2.不要排空任何有变色或腐蚀迹象或有异常气味的七氟醚蒸发器。移动蒸发器进行储存时,使用防护手套、护目镜并遵循一般化学处理安全指南。 3.如果蒸发器内没有变色或腐蚀的迹象或异常气味,清空蒸发器并按照相关说明执行干燥运行,说明包含在信函和使用说明中。 4.请联系Getinge公司在当地的代表或向销售支持发送电子邮件(CSalesSupport@getinge.com)请求退货授权(RMA)和装运说明,以退回空的受影响产品。 5.如有必要,请向Getinge公司代表咨询与有变色/腐蚀迹象的蒸发器退货相关的具体说明。 6.知转发给任何需要了解此信息的人,无论是在本单位还是在受影响设备被转移到的任何其他场所。 7.完成并签署信函所附的紧急医疗器械召回回复表,即使没有受影响的产品。 (美国FDA网站)
美国FDA发布关于BD公司因存在加载过时自动化编程请求风险召回Alaris输液泵系统的警示信息
发布日期:2025年3月20日 召回级别:此次召回涉及纠正器械的软件,不涉及从使用单位或经营企业撤回上述器械。FDA已将此次召回确定为最严重的类型。如果您不按照更新的说明继续使用此器械,可能会导致严重伤害或死亡。 召回产品:
产品名称:BD Alaris系统管理器和BD护理协调引擎(CCE)输液适配器软件 UDI编码: BD Alaris系统管理器软件: 10885403960123, 10885403519666, 10885403960116/9601 BD CCE输液适配器软件: 10885403510472 软件版本: BD Alaris系统管理器:软件v12.5.1或v12.5.2,和较早版本 (4.33, 12.0.1, 12.0.2, 12.1, 12.1.2, 12.3) 产品用途: BD Alaris 系统是一种输液泵和监测系统,可用于持续(连续输注)或间歇(重复输注)输送液体,如药物、红细胞或其他血液制品。该系统可通过静脉(IV)或动脉(IA)输注,也可用于皮下(SC)、硬膜外(Epidural)或体液腔内输注,适用于成人、儿童和婴儿。 BD Alaris 系统管理器软件连接至 Alaris 系统计算机,以支持各组件协同工作。护理协调引擎(CCE)输液适配器软件使 BD Alaris 系统能够与医院的电子病历(EMR)系统对接,并支持自动化的泵编程工作流程。 召回原因: BD公司正在对 BD Alaris 系统管理器和 BD 护理协调引擎(CCE)输液适配器的软件进行更正。此前,BD 收到报告称,使用系统管理器软件且连接 CCE 输液适配器的客户可能会遇到系统响应延迟和自动化编程请求(APR)积压的问题。 在延迟发生后,过时的 APR 可能会被上传到 Alaris 计算机,并可能导致输液速率、剂量或体积参数与当前输液方案不一致。如果用户接受了过时的速率/剂量更改,患者可能会接受不准确的治疗,包括输液过量(过度输注)或不足(输液不足)。 受影响产品的使用可能会导致严重的不良健康后果,包括因输液泵突然停止或输液量异常(过量或不足)而引发的并发症,甚至死亡。 目前尚未收到相关伤害或死亡报告。 召回措施: 2025年2月18日,Becton Dickinson and Company(BD)及其子公司 CareFusion 向所有受影响的客户发送了紧急医疗设备产品更正通知,建议采取下述措施,以防止因软件问题导致输液参数错误,保障患者安全:
(美国FDA网站)
|





|