

|
医疗器械警戒快讯 2024年第12期(总第214期)
发布日期:2025-01-03
医疗器械警戒快讯
(总第214期) 内容提要
澳大利亚TGA发布关于Accuray公司因标准治疗床缺陷问题召回CyberKnife治疗实施系统的警示信息 发布日期:2024年11月28日 召回级别:II级 召回编号:RC-2024-RN-01068-1 召回产品:使用标准治疗床的CyberKnife治疗实施系统 产品名称/描述: 产品代码:0660000 序列号:C0345、C0447 ARTG ID:155887(Emergo Asia Pacific Pty Ltd T/a Emergo Australia-加速器系统,线性) 责任主体:Emergo Asia Pacific Pty Ltd T/a Emergo Australia 召回原因:Accuray公司发现CyberKnife标准治疗床(STC)存在问题。在极少数情况下,卡环会从内部结构的轴上部分或完全脱落。临床使用时,如果内部结构断开,就会出现不受控制的旋转,直到被地面或治疗床支柱的间距所阻挡才会停止。出现这种情况时,患者有可能从STC上掉落。 召回措施:Accuracy公司将检查STC,并且无论状况如何,都会更换现有受影响STC固定架上的卡环。该问题可能在使用STC期间随时出现并影响临床使用。客户可以继续在临床上使用CyberKnife系统。客户应确保在放置、重新定位和离开STC时照顾患者。 (澳大利亚TGA网站)
美国FDA发布关于Elekta公司因活检针内部微小不锈钢碎片风险召回一次性活检针包的警示信息 发布日期:2024年11月29日 召回级别:FDA将此识别为最严重的召回类型,继续使用这些产品可能造成严重伤害或者死亡。此次召回涉及从使用机构或销售机构撤回相关产品。 受影响的产品: 产品用途:一次性活检针包在神经外科中用于精确的脑组织取样。它旨在与Leksell立体定向系统一起使用,允许外科医生在大脑中的目标位置准确地获取组织样本。 召回原因: 由于活检针内部存在微小碎片的风险,Elekta公司正在召回与Leksell立体定位系统一起使用的一次性活检针包。碎片中的材料是不锈钢,与活检针的材料相同。在活检针的外部没有发现碎片。活检针的无菌性没有受到影响。 使用受影响的产品可能会导致严重的不良健康后果,包括碎片进入脑组织和死亡。 目前还没有收到伤害和死亡的报告。 采取措施: 2024年9月25日,Elekta公司向所有受影响的客户发送了紧急重要现场安全通知,建议采取以下措施: (美国FDA网站)
美国FDA发布关于Cardinal Health公司因与无针注射器不兼容的风险而召回某些Monoject U-100 mL鲁尔锁 胰岛素注射器(带尖端盖软包装)的警示信息 发布日期:2024年12月4日 召回级别:FDA将此识别为最严重的召回类型,继续使用这些产品可能造成严重伤害或者死亡。此次召回涉及从使用机构或销售机构撤回相关产品。 受影响的产品: 产品用途:Monoject U-100 mL鲁尔锁胰岛素注射器,带尖端盖软包装,用于使用鲁尔兼容注射器在皮肤下(皮下)或通过血管(静脉内)注射胰岛素,以治疗血液中过量的钾(急性高钾血症)。 召回原因: 由于与无针静脉注射连接器不兼容,Cardinal Health公司正在召回部分批次的Monoject U-100 1ml带尖端盖软包装鲁尔锁胰岛素注射器。如果受影响的产品用于通过无针连接器进行静脉注射胰岛素,由于不是所有的药物都会离开注射器,可能会增加患者无法接受足够剂量胰岛素的风险。 使用受影响的产品可能会导致严重的不良健康后果,包括高血糖或低血糖、脂肪在体内分解过快(糖尿病酮症酸中毒)和死亡。 目前没有收到伤害和死亡的报告。 采取措施: 2024年9月25日,Cardinal Health公司向所有受影响的客户发送了一封紧急医疗器械召回函,建议采取以下行动: 无论是否有受影响的产品,请通过传真(614-652-9648)或电子邮件(GMB-FieldCorrectiveAction@cardinalhealth.com)将信函附带的确认表返回。 (美国FDA网站)
美国FDA发布关于Fresenius Kabi USA公司Ivenix输液泵气动阀门故障报警风险的警示信息 本通知是“加强医疗器械召回工作的沟通试点”的一部分内容。FDA已经意识到一个潜在的高风险器械问题。FDA将持续向公众通报,并在获得重大新信息时更新网站公布的相关信息。 发布日期:2024年12月11日 受影响的产品:Ivenix大容量输液泵(LVP-0004),UDI 00811505030320 产品序列号:详见FDA网站https://www.fda.gov/medical-devices/medical-device-recalls/early-alert-infusion-pump-issue-fresenius-kabi-usa FDA已获悉,Fresenius Kabi USA公司已向受影响的医疗服务提供者发出通知,指出部分Ivenix LVP需要停止使用以进行修理。 警示原因:Fresenius Kabi USA公司报告称,部分Ivenix LVP中安装的气动阀门存在较高的非恢复性泵故障报警风险。所有带受影响阀门的产品应按照下述“应采取的措施”内容,停止使用,进行评估并返回Fresenius Kabi公司进行处理。 泵故障报警正在按预期工作,如果发生故障,将发出信号指示何时采取行动。 公司尚未收到与此问题相关的任何伤害和死亡报告。 采取措施: 2024年12月5日,Fresenius Kabi USA公司向所有受影响的客户发出通知,建议在受影响的LVP返回进行气动阀门修复之前,采取以下措施:
(美国FDA网站)
美国FDA发布Hologic公司因并发症风险召回BioZorb 3D生物可吸收标志物的警示信息 发布日期:2024年12月18日 召回级别:FDA将此识别为最严重的召回类型,继续使用这些产品可能造成严重伤害或者死亡。此次召回涉及从使用机构或销售机构撤回相关产品。 受影响的产品: 产品用途:Hologic公司(前身是Focal Therapeutics公司)制造的BioZorb标志物是一种可植入的放射学标记,用于标记软组织(如乳腺组织)以进行未来的医疗程序。 该产品有两个组件:一个是由钛金属制成的永久性组件,一个是由塑料材料制成的可吸收组件,将在1年或更长时间后在体内被吸收。产品为一次性使用。 召回原因:Hologic公司由于植入产品相关的并发症和不良事件正在召回Biozorb标志物。 使用受影响产品可能会导致严重的不良健康后果,包括疼痛、感染、皮疹、产品从放置位置移动(迁移)、产品穿过皮肤(侵蚀)、皮肤下液体积聚(血肿)、其他并发症和/或额外的治疗以及死亡。 已经收到252份伤害报告。目前没有收到死亡报告。 采取措施: 不要使用BioZorb标志物和BioZorb LP标志物。 监测植入BioZorb标志物的患者是否有任何不良事件的迹象。 2024年10月24日,Hologic公司向所有受影响的客户发送了紧急医疗器械召回信,建议采取以下措施: 患者 医疗服务提供者 相关机构 全部受影响产品的清单
(美国FDA网站) 美国FDA发布关于Trokamed公司肾镜管鞘不可用于抽吸和冲洗的警示信息 本通知是“加强医疗器械召回工作的沟通试点”的一部分内容。FDA已经意识到一个潜在的高风险器械问题。FDA将持续向公众通报,并在获得重大新信息时更新网站公布的相关信息。 发布日期:2024年12月20日 受影响的产品:Trokamed公司Mini PCNL肾镜管鞘 FDA了解到Trokamed公司已向受影响的医疗保健提供者发出一封信,表明由于潜在的高风险器械问题,已更新某些内窥镜管鞘使用说明。 涉及以下型号所有批次的Mini PCNL肾镜管鞘产品:
产品用途:Mini PCNL肾镜管鞘是肾镜附件套件的一部分,用于包括检查肾脏和去除肾结石或其他阻塞物在内的微创手术。管鞘是一种短期使用的可重复使用、手术侵入性器械,被设计用于将仪器、内镜和液体带到手术部位。 警示原因:该器械之前随附的使用说明没有明确提示管鞘不可用于抽吸和冲洗。如果该器械用于抽吸或冲洗,则可能会因血液、组织和肾结石碎片堵塞而减少通过管轴的流出量,这可能会迅速在肾脏内积聚压力,从而导致肾脏破裂。 Trokamed公司已报告了1份与此问题相关的死亡报告。 采取措施: 2024年11月26日,Trokamed公司向所有受影响的客户发送了一份紧急现场安全通知,建议采取以下措施: (美国FDA网站)
|
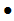 澳大利亚TGA发布关于Accuray公司因标准治疗床缺陷问题召回CyberKnife治疗实施系统的警示信息
澳大利亚TGA发布关于Accuray公司因标准治疗床缺陷问题召回CyberKnife治疗实施系统的警示信息


|