

|
医疗器械警戒快讯 2024年第9期(总第211期)
发布日期:2024-09-30
医疗器械警戒快讯
内容提要
加拿大Health Canada发布关于GE Healthcare公司因软件问题召回PACS系统的警示信息 发布日期:2024年8月30日 召回级别:II级 召回产品:具体型号和批次见下表:
召回发起时间:2024年8月22日 召回原因:GE Healthcare公司意识到一个问题,即当Centricity PACS达到唯一图像标识符(Ckeys)的最大数量时,将无法进行图像存储。如果出现这种情况,就会导致只能获取到部分检查图像。万一部分检查图像未被注意到,则可能导致误诊。Centricity PACS能够唯一识别约21亿张图像(例如,相当于2100万次检查,每次检查100张图像)。但是,当达到唯一图像标识符的最大数量时,用户不会收到任何通知。 如需更多信息,请联系制造商。 (加拿大Health Canada网站)
美国FDA发布关于Medtronic公司因电池过热和爆炸风险召回部分McGrath MAC视频喉镜的警示信息 发布日期:2024年9月5日 召回级别:I级,是最严重的召回类型,使用这些产品可能造成严重伤害或者死亡。此次召回涉及从使用机构或销售机构撤回相关产品。此次召回还包括更新相关产品其他型号的使用说明,但不包括将其从使用机构或销售地点撤回。 图:McGrath MAC视频喉镜3.6v电池使用日期和序列号标签 涉及召回的产品: 涉及使用说明更新的产品: 产品用途:McGrath MAC和MAC EMS视频喉镜用于帮助医护人员在医疗过程中观察气管。该产品包括一个光源和一个微型摄像机用于在喉镜检查中观察喉腔(喉)。 召回产品和更新使用说明的原因: Medtronic公司正在召回某些产品,并更新另一些产品的使用说明,原因是电池可能会耗尽到设计阈值以下。如果发生这种情况,会增加电池不稳定的风险,并可能导致电池温度升高和潜在的爆炸。 使用受影响的产品可能会导致严重的不良健康后果,包括烧伤、割伤(裂伤)、疤痕和其他组织损伤、牙齿脱落、眼睛损伤、听力受损(损伤或声震)或耳鸣(耳鸣)、呼吸衰竭、身体缺氧(缺氧)、疤痕和死亡。 有1份伤害报告,无死亡报告。 采取措施: 对于McGrath MAC和MAC EMS视频喉镜 对于McGrath MAC视频喉镜(Next Generation) 2024年7月,Medtronic公司向所有受影响的客户发送了一封紧急医疗设备移除和纠正信函,建议采取以下额外措施: 对于McGrath MAC和MAC EMS视频喉镜 对于McGrath MAC视频喉镜(Next Generation) 适用于所有产品(无需考虑撤回或更新IFU) (美国FDA网站)
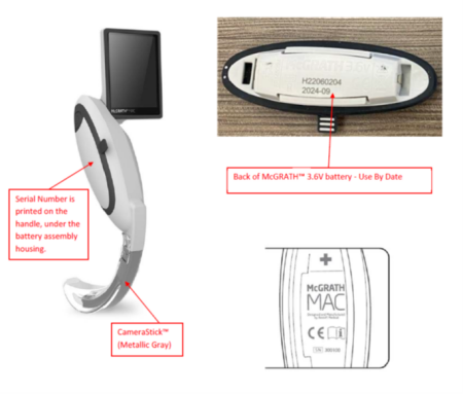
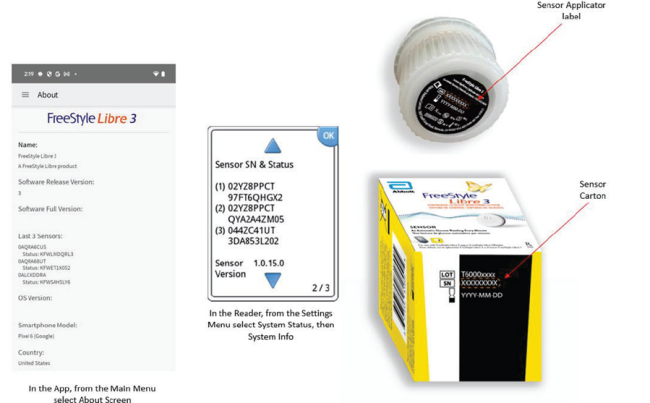
美国FDA发布关于Abbott Diabetes Care公司因不准确高葡萄糖读数风险召回部分FreeStyle Libre 3传感器的警示信息 发布日期:2024年9月5日 召回级别:I级,是最严重的召回类型,使用这些产品可能造成严重伤害或者死亡。此次召回涉及从使用机构或销售机构撤回相关产品,但不适用于FreeStyle Libre3阅读器或应用程序。 图: app屏幕图片,阅读器设置,传感器标签,传感器纸箱 召回产品: 产品用途:FreeStyle Libre 3连续血糖监测系统旨在提供对血糖水平的连续监测。它通过发现趋势和跟踪葡萄糖水平模式来帮助用户管理糖尿病,以便根据需要调整治疗。它适用于单个患者并需要处方。 召回原因: Abbott Diabetes Care公司在发现少量FreeStyle Libre 3传感器可能提供不准确的高葡萄糖读数后,正在召回这些FreeStyle Libre 3传感器,如果未被检测到上述问题,可能会对糖尿病患者造成潜在的健康风险。 使用受影响的产品可能会导致严重的不良健康后果,包括严重低血糖(低血糖症),这可导致中枢神经系统问题、意识丧失、癫痫发作、昏迷、永久性脑损伤和死亡。 有2份伤害报告,还没有死亡报告。 采取措施: 2024年7月24日,Abbott Diabetes Care公司向所有受影响的客户发送了紧急医疗器械召回通知,建议采取以下措施: (美国FDA网站)
美国FDA发布关于Fresenius Kabi公司因制造缺陷召回部分Ivenix输注系统LVP的警示信息 发布日期:2024年9月16日 召回级别:I级,是最严重的召回类型,使用这些产品可能造成严重伤害或者死亡。此次召回涉及从使用机构或销售机构撤回相关产品。 召回产品: 产品用途: Ivenix大容量泵(LVP)是Ivenix输注系统的三个主要组件之一。它是一种利用气压来精确控制输入患者体内的液体流量的泵。该输注系统用于医院和门诊中心,通过静脉注射、动脉注射、脊柱注射或皮下注射等不同途径,精准地为患者输液。它适用于成人、儿科和新生婴儿。 Ivenix大容量泵仅与特定的无菌、一次性使用的给药装置兼容,包括主给药装置,双通道,低吸附,无针端口,Y型三通。 召回原因: Fresenius Kabi公司在发现可能导致药物流速失控的制造缺陷后,正在召回部分的Ivenix LVP主给药装置。 使用受影响的产品可能会导致严重的不良健康后果,包括用药过量和死亡。 已收到2份伤害报告。没有收到死亡报告。 采取措施: 此次召回仅限于特定批次:3010538。其他批次不受影响。 2024年8月2日,Fresenius Kabi公司向所有受影响的客户发送了一封紧急主动召回信函,建议采取以下措施: (美国FDA网站)
美国FDA发布关于Zimmer Biomet CPT髋关节系统股骨柄相关大腿骨折风险增加的安全信息通报 发布日期:2024年9月17日 美国食品药品监督管理局(FDA)提醒患者、护理人员、医务人员和医疗机构注意,使用Zimmer Biomet CPT髋关节系统12/14颈锥度股骨柄(CPT髋关节系统)会增加手术后大腿骨折(术后假体周围股骨骨折)的风险。 2024年7月2日,由于大腿骨折的风险增加,Zimmer Biomet公司启动了主动召回,以更新CPT髋关节系统的使用说明。该制造商还宣布,计划在2024年12月之前逐步停止该产品的销售。然而,最近研究发现与类似设计的髋关节置换产品相比,该产品发生大腿骨折的风险更高,并且如果发生骨折,可能需要手术干预,FDA因此对新患者继续植入CPT髋关节系统表示担忧。FDA正在与制造商合作解决这些问题。 对患者和护理人员的建议 对医务人员和医疗机构的建议 产品用途:CPT髋关节系统12/14颈锥度股骨柄用于髋关节置换术,是一种由钴铬合金制成的抛光锥形滑动(PTS)型柄。 大腿骨折的风险增加 最近的研究发现,与类似设计的髋关节置换产品相比,使用CPT髋关节系统手术后大腿骨折的风险更高。根据英国药品和保健产品监管局(MHRA)于2024年9月4日发布的信息,一份目前尚未发表的对英国最常见植入PTS髋关节柄的分析表明,CPT髋关节系统患者的大腿骨周围骨折风险最高,约为1.4%,而类似的PTS髋关节柄的骨折率约为0.6%至1%。 FDA采取的措施: FDA将继续与Zimmer Biomet公司合作,帮助确保患者、护理人员、医务人员和医疗机构意识到CPT髋关节系统会增加大腿骨折的风险。 FDA还将继续监测产品性能、大腿骨折报告,并与Zimmer Biomet公司合作,根据需要确定其他缓解策略。 FDA将继续与国际监管机构合作,审查数据并进一步评估产品性能。 如有重要的新信息,FDA将随时通知公众。 (美国FDA网站)
美国FDA发布关于Smiths Medical公司因制造缺陷召回部分Bivona新生儿/儿童和成人气管造口管的警示信息 发布日期:2024年9月18日 召回产品: 图:因制造缺陷导致断裂的产品 产品用途:Bivona新生儿/儿童和成人气管造口管用于为气管造口患者提供长达29天的直接气道通道。一些气管造口管可以在同一患者上重新处理和重复使用多次。 召回原因:Smiths Medical公司正在召回特定批次的Bivona新生儿/儿童和成人气管造口管,原因是制造缺陷可能导致产品的固定法兰撕裂。 使用受影响的产品可能会导致严重的不良健康后果,包括缺乏适当的通气、失去受保护的气道和死亡。 据报道有35份伤害报告,有2份死亡报告。 召回措施:不要使用受影响批号的Bivona气管造口管并处理掉相关产品。 2024年5月29日,Smiths Medical公司向所有受影响的客户发送了一份紧急医疗器械通知函,建议采取以下行动: (美国FDA网站)
|
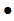 加拿大Health Canada发布关于GE Healthcare公司因软件问题召回PACS系统的警示信息
加拿大Health Canada发布关于GE Healthcare公司因软件问题召回PACS系统的警示信息


|