

|
医疗器械警戒快讯 2024年第8期(总第210期)
发布日期:2024-08-30
医疗器械警戒快讯
内容提要 加拿大Health Canada发布关于Karl Storz公司因标签和包装问题召回内窥镜的警示信息 澳大利亚TGA发布关于Smiths Medical澳大利亚有限公司因制造缺陷问题召回Portex Blue Line 硅化 PVC 气管切开插管的警示信息 美国FDA发布关于Abiomed公司因质量检查失败问题召回带有SmartAssist系统的Impella CP心脏泵的警示信息 美国FDA发布关于Smiths Medical公司CADD-Solis便携式输液泵软件纠正的警示信息 美国FDA发布关于Defibtech公司因存在停止按压问题召回RMU-2000 ARM XR胸部按压装置的警示信息 澳大利亚TGA发布关于Fisher & Paykel Healthcare公司因可能导致缺氧问题召回Airvo 3呼吸支持设备的警示信息
加拿大Health Canada发布关于Karl Storz公司因标签和包装问题召回内窥镜的警示信息
发布日期:2024年8月1日 召回级别:II级 召回产品:具体型号和批次见下表:
制造商:Karl Storz Se & Co. Kg 召回发起时间:2024年7月25日 召回原因:上表所列器械标识符目前来自 Karl Storz在美国的另一家子公司。作为其 510(k) 更新的一部分,其 IFU(使用说明) 已被更改。作为获准的再处理方式,这些器械的 IFU 以前包括除手工清洗和预真空蒸汽以外的清洗方法;现在IFU已更新为今后只包括这两种方法;目前存在未获批准的再处理方式可能被用于再处理的风险。 如需更多信息,请联系制造商。 (加拿大Health Canada网站)
澳大利亚TGA发布关于Smiths Medical澳大利亚有限公司因制造缺陷问题召回Portex Blue Line 硅化 PVC 气管切开插管的警示信息
发布日期:2024年8月1日 召回级别:Ⅰ级 召回编号:RC-2024-RN-00649-1 召回产品:Portex Blue Line 硅化PVC气管切开插管 产品批号:多个批号 产品注册号:ARTG 340372(Smiths Medical Australasia Pty Ltd – 插管,气管切开术,一次性使用) 召回原因: 这是对2024年5月31日来文(RC-2024-RN-00429-1)的更新。Smiths Medical确定了其他可能受到原始通知中确定的制造缺陷影响的产品和批次。这些产品于2019年5月至2023年2月期间分销。 特定批次的Blue Line气管切开术产品的固定翼可能会因制造缺陷而撕裂。 如果插管上的固定翼撕裂或破损,插管可能无法在气管中保持原位。这可能导致气管切开术移位或脱管。任何一种情况都可能导致无法正常通气或保护气道。任何一种情况都可能导致灾难性的不良事件。 迄今为止,Smiths Medical已收到35份与此问题相关的严重伤害报告和1份死亡报告。 召回措施:用户应检查本机构内所有库存,以确定是否存在受影响的批号,并停止使用。按照每个机构的废弃流程,废弃所有受影响的产品。如果机构暂时无法立即废弃,应将产品隔离直至被处置。 (澳大利亚TGA网站)
美国FDA发布关于Abiomed公司因质量检查失败问题召回带有SmartAssist系统的Impella CP心脏泵的警示信息
发布日期:2024年8月12日 召回级别:I级,是最严重的召回类型,使用这些产品可能造成严重伤害或者死亡。此次召回涉及从使用方或销售方撤回相关产品。 召回产品:
产品用途: 带有SmartAssist的Impella CP心脏泵用于在高风险的基于导管的手术,也被称为经皮冠状动脉介入治疗(PCI)时,对心脏动力泵区(心室)提供短期支持。 带有SmartAssist的Impella CP心脏泵适用于以下情况:在严重心脏病(急性心肌梗塞)发作48小时内发生的心原性休克、当心脏因心肌症无法正常工作而进行体外循环心脏手术。 Impella疗法旨在减少心脏左心室的工作,并为循环系统提供支持,以便心脏有恢复时间。 召回原因: Abiomed公司在发现某批次中的9个心脏泵没有通过检查,但却被不经意地发给客户之后,正在召回某些带有SmartAssist的Impella CP心脏泵。使用受影响的心脏泵可能会导致意外停泵或释放有潜在危害的颗粒。 使用受影响的产品可能会导致严重的不良健康后果,包括心脏和血管(心血管)并发症,如中风、低血压、红细胞损伤(溶血)、出血、心包积液(心脏压塞)、心脏病发作(心肌梗死)、需要额外的外科手术或死亡。 没有收到伤亡报告。被召回的产品都没有用于病人护理。 召回措施: 2024年5月31日,Abiomed公司向客户发送了一封紧急医疗器械自愿召回函,建议采取以下措施:
(美国FDA网站)
美国FDA发布关于Smiths Medical公司CADD-Solis便携式输液泵软件纠正的警示信息
发布日期:2024年8月6日 召回级别:此次召回涉及纠正某些特定器械,不涉及将其从使用或销售的地方移除。FDA已将此次召回确定为最严重的类型。如果您继续使用此设备而不进行纠正,可能会导致严重伤害或死亡。 召回产品: CADD-Solis便携式输液泵 CADD-Solis v.4.2之前的各种软件版本 CADD-Solis VIP v.1.6之前的各种软件版本 v.2.6之前的PharmGuard服务器软件版本 UDI/型号:详见下图
产品用途: CADD-Solis和CADD-Solis-VIP输液泵适用于以下用途: 用于血管(静脉内)输注,包括动脉(动脉内)、皮肤下(皮下)、腹部(腹腔内)、靠近神经、手术部位(术中)、脊柱椎骨之间(硬膜外腔或蛛网膜下腔)。 需要持续输注速率和/或间歇性大剂量(推注)和/或患者控制的按需剂量的治疗。 纠正原因: 由于在没有最新软件版本的情况下使用这些输液泵时可能会出现多种问题,Smiths Medical公司正在纠正CADD Solis和CADD Solis-VIP便携式输液泵的软件。v4.3之前的软件版本可能会出现潜在问题,包括:
使用受影响的产品可能会导致严重的不良健康后果,如延迟、中断、治疗不足或过度或死亡。 目前有一例伤害报告,没有死亡报告。 纠正措施: 确保所有泵上安装了最新的CADD软件。 Smiths Medical通过之前的软件更新已经纠正了许多问题,这些纠正被延续到所有后续软件版本中。 2024年2月27日,Smiths Medical向所有受影响的客户发送了一封紧急医疗器械更正函,建议采取以下行动: 对于用户 找到受影响的泵; 确保所有用户或潜在用户都知道通知和更正; 10天内填写并返回随信附上的回复表至smithsmedical3563@sedgwick.com,即使没有受影响的产品,也要填写。 对于分销商 向可能收到潜在受影响产品的任何客户发出通知。 要求他们填写回复表并将其寄回smithsmedical3563@sedgwick.com. (美国FDA网站)
美国FDA发布关于Defibtech公司因存在停止按压问题召回RMU-2000 ARM XR胸部按压装置的警示信息
发布日期:2024年8月22日 召回级别:I级 召回产品:RMU-2000 ARM XR胸部按压装置 UDI:UDI-DI: 00815098020812, 10815098020819 公司发起日期:2024年7月12日
产品用途:RMU-2000 ARM XR胸部按压装置用于对心脏突然停止并且血液在全身不循环的成人提供胸外按压。 召回原因:Defibtech公司正在召回RMU-2000 ARM XR胸部按压装置,原因是该装置的电机存在问题,可能导致其停止按压。 使用受影响产品可能会造成严重的不良健康后果,包括患者受伤、治疗延误和由于一段时间没有按压使氧气在全身循环而导致的死亡。 据报道,已有一人受伤,一人死亡。 召回措施: 请勿使用受影响的Defibtech RMU-2000 ARM XR胸部按压装置。 2024年7月12日,Defibtech公司向所有受影响的客户发送了一封紧急医疗器械安全解除函,建议采取以下行动: 识别并隔离受影响的单位。 如果设备被进一步转移或分发,请确保收件人知道此通知或向Defibtech公司提供联系信息。 Defibtech公司将联系客户安排产品退货,设备将免费退款、维修或更换。 (美国FDA网站)
澳大利亚TGA发布关于Fisher&Paykel Healthcare公司因可能导致缺氧问题召回Airvo 3呼吸支持设备的警示信息
发布日期:2024年8月21日 召回级别:I级 召回产品:Airvo 3 NIV呼吸支持设备 召回原因:当使用高压氧(HPO)设置并运行1.5.1或更早版本软件的Airvo 3 NIV设备发生3.2.2流量校准报警时,该设备将仅输送室内空气。如果发生这种情况,患者可能会经历氧饱和度降低,从而导致缺氧。 召回措施: 收到确认表后,Fisher&Paykel Healthcare将与客户联系,安排将受影响的产品更新到软件版本1.5.2。 在软件更新之前,客户可以继续使用该设备。使用设备时,客户必须遵循所有说明,包括用户手册中的警告和注意事项,特别是第1节和第2节中的警告和注意事项。如果出现3.2.2报警,客户应遵循屏幕上的说明。 (澳大利亚TGA网站)
|

 产品名称:带SmartAssist的Abiomed Impella CP
产品名称:带SmartAssist的Abiomed Impella CP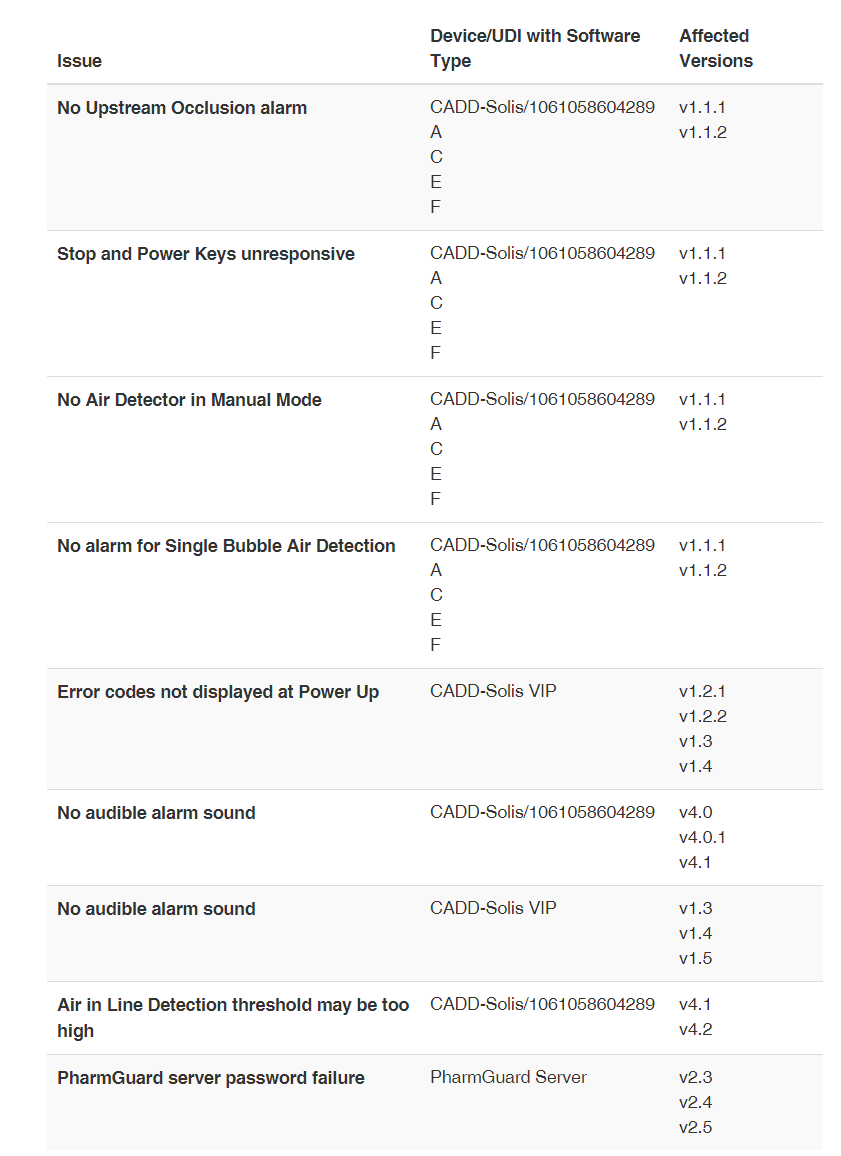


|