|

|
国家药品不良反应监测中心
国家药品监督管理局药品评价中心
http://www.cdr-adr.org.cn
|
加拿大Health Canada发布关于GE Healthcare公司召回Ic9-Rs超声探头的的警示信息
发布日期:2024年1月24日
召回级别:Ⅱ级
召回产品:Ic9-Rs超声探头,型号H48691PJ(IC9-RS),超过10批次,具体批号请咨询生产商。
制造商:GE Healthcare Austria公司
召回发起时间:2024年1月9日
召回原因:由于Ic9-Rs腔内探头出现双重图像伪影,GE Healthcare Austria公司正在召回特定型号的Ic9-Rs超声探头。如果需要更多相关信息,请与生产商联系。
(加拿大Health Canada网站)
美国FDA发布关于Cardinal Health公司因与注射泵不兼容召回一次性使用注射器的警示信息
发布日期:2024年2月2日
召回级别:Ⅰ级,是最严重的召回类型,使用这些产品可能造成严重伤害或者死亡。
召回产品:Cardinal Health Monoject一次性使用Luer-lock注射器1,6,12,20,35和60mL
 Monoject 1mL Luer-lock尖软包装结核菌素注射器
Monoject 1mL Luer-lock尖软包装结核菌素注射器
 Monoject 6mL Luer-lock尖软包装注射器
Monoject 6mL Luer-lock尖软包装注射器
 Monoject 12mL Luer-lock尖软包装注射器
Monoject 12mL Luer-lock尖软包装注射器
 Monoject 20mL Luer-lock尖软包装注射器
Monoject 20mL Luer-lock尖软包装注射器
 Monoject 35mL Luer-lock尖软包装注射器
Monoject 35mL Luer-lock尖软包装注射器
 Monoject 60mL Luer-lock尖软包装注射器
Monoject 60mL Luer-lock尖软包装注射器
产品编号:详见召回数据库信息
https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfRES/res.cfm?id=203389
分销日期:2023年6月1日至2023年8月31日
在美国召回数量:32,433,200
召回发起日期:2023年9月19日
产品用途:Cardinal Health公司Monoject一次性使用注射器用于向体内注射液体或从体内取出液体。当与注射泵一起使用时,Monoject一次性使用注射器用于装载液体或药物并放入注射泵中。
注射泵用于为患者输送液体、药物和血液制品等。
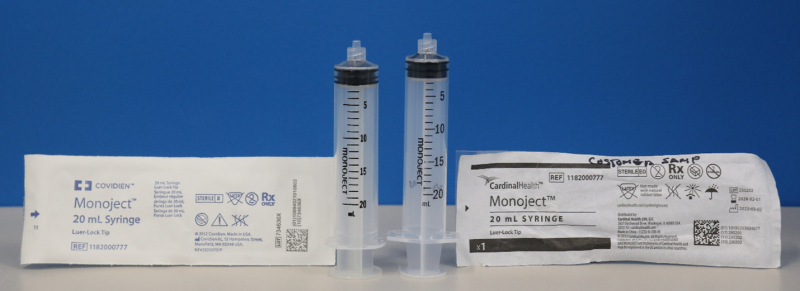
召回原因:2023年6月,Cardinal Health公司开始分销品牌为“Cardinal Health Monoject syringes”(Cardinal Health Monoject注射器)的Monoject注射器。这些新注射器与之前品牌为“Covidien Monojects syringes”(Covidien Monojects注射器)不同,因为它们的尺寸不同。
如上图所示,左边的Covidien Monoject注射器与右边的Cardinal Health Monoject注射器具有不同的尺寸。
受影响的Cardinal Health Monoject注射器(见上图右侧)不应与注射泵一起使用。与注射泵一起使用时,受影响的Cardinal Health Monoject注射器的尺寸变化可能会导致注射泵性能出现问题,如过量、剂量不足、治疗延迟和阻塞警报延迟。如上图所示,左边的Covidien Monoject注射器与右边的Cardinal Health Monoject注射器具有不同的尺寸。
Cardinal Health公司已经收到了15份因注射泵无法识别注射器而导致治疗延迟的报告,以及13份容量/速率不准确的报告,包括一些伤害报告。Cardinal Health公司尚未收到任何患者死亡报告。
受影响人群:
 与注射泵一起使用Cardinal Health Monoject一次性使用注射器的人员
与注射泵一起使用Cardinal Health Monoject一次性使用注射器的人员
 与注射泵一起使用Cardinal Health Monoject一次性使用注射器的医护人员
与注射泵一起使用Cardinal Health Monoject一次性使用注射器的医护人员
采取措施:
 请勿将受影响的Cardinal Health Monoject注射器与注射泵一起使用。有关受影响产品的列表,请参阅下表。
请勿将受影响的Cardinal Health Monoject注射器与注射泵一起使用。有关受影响产品的列表,请参阅下表。
|
产品代码
|
产品描述
|
UDI
|
产品批号
|
|
1180100777
|
Monoject 1 mL Tuberculin Syringe Luer-Lock Tip Soft Pack
|
10192253034530 - each
20192253034537 - box
50192253034538 - case
|
221201, 221202, 221203,
230201, 230202, 230203,
230204, 230205, 230601
|
|
1180600777
|
Monoject 6 mL Syringe Luer-Lock Tip Soft Pack
|
10192253034608- each
20192253034605- box
50192253034606- case
|
221201, 221202, 221203,
221204, 221205, 230201,
230202, 230203, 230204,
230205, 230206, 230207
|
|
1181200777T
|
Monoject 12 mL Syringe Luer-Lock Tip Soft Pack
|
10192253025811-each
20192253025818-box
50192253025819-case
|
221101, 221102, 221103,
21104
|
|
1182000777
|
Monoject 20 mL Syringe Luer-Lock Tip Soft Pack
|
10192253034677-each
20192253034674-box
50192253034675-case
|
221201, 221202, 221203,
221204, 221205, 230201,
230202, 230203, 230204,
230205, 230206
|
|
1183500777
|
Monoject 35 mL Syringe Luer-Lock Tip Soft Pack
|
10192253034691-each
20192253034698-box
50192253034699-case
|
221201, 230201, 230601,
230602
|
|
1186000777T
|
Monoject 60 mL Syringe Luer-Lock Tip Soft Pack
|
10192253025835-each
20192253025832-box
50192253025833-case
|
221101, 230601
|
 您可以继续使用Covidien Monoject注射器和注射泵。
您可以继续使用Covidien Monoject注射器和注射泵。
2023年9月20日,Cardinal Health公司向所有受影响的客户发送了一封紧急医疗器械产品更正函。
信函要求客户:
 查看库存中受影响的产品代码和批次。产品代码和批次如上表所示。
查看库存中受影响的产品代码和批次。产品代码和批次如上表所示。
 与所有使用Cardinal Health Monoject Luer-Lock尖注射器(1、6、12、20、35和60mL)的人员沟通,提示这些注射器不应与注射泵一起使用。
与所有使用Cardinal Health Monoject Luer-Lock尖注射器(1、6、12、20、35和60mL)的人员沟通,提示这些注射器不应与注射泵一起使用。
 将此通知的副本张贴在存放产品的储藏室中。
将此通知的副本张贴在存放产品的储藏室中。
 通知您可能已向其分发/转发受影响产品或将向其发送产品的任何客户此医疗器械产品更正函内容,并共享此通知的副本。
通知您可能已向其分发/转发受影响产品或将向其发送产品的任何客户此医疗器械产品更正函内容,并共享此通知的副本。
 无论您是否拥有受影响的产品,请将提供的确认表格发送传真至614-652-9648或电子邮件至GMB-FieldCorrectiveAction@cardinalhealth.com
无论您是否拥有受影响的产品,请将提供的确认表格发送传真至614-652-9648或电子邮件至GMB-FieldCorrectiveAction@cardinalhealth.com
(美国FDA网站)
美国FDA发布关于Percussionaire公司因超压召回高频输送Phasitron呼吸回路套件的警示信息
发布日期:2024年2月6日
召回级别:I级,是最严重的召回类型,使用这些产品可能造成严重伤害或者死亡。
召回产品:
 产品名称:高频输送Phasitron呼吸回路套件(A50605-D)
产品名称:高频输送Phasitron呼吸回路套件(A50605-D)
 产品代码:请参阅调用数据库条目
产品代码:请参阅调用数据库条目
 发起日期:2023年6月9日至2023年12月1日
发起日期:2023年6月9日至2023年12月1日
 美国召回数量:2145台
美国召回数量:2145台
 召回发起日期:2023年12月12日
召回发起日期:2023年12月12日
产品用途:
高频输送Phasitron呼吸回路套件用于患者的持续通气。该套件旨在与冲击式高频冲击通气(HFPV)系统配合使用,用于在医院内或入院前提供紧急护理。该套件也可用于医院内外的运输。
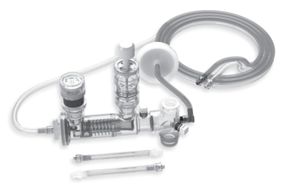
召回原因:
由于产品缺陷导致超压,Percussionaire公司正在召回其高频输送Phasitron呼吸回路套件。Phasitron套件有两部分在制造过程中必须压在一起。如果一个零件角度不正确并受到挤压,该零件可能会变形。这导致PEEP(呼气末正压通气)阀门卡在关闭位置。当阀门被卡住时,没有办法手动释放阀门。
使用受影响的Phasitron套件可能会导致严重的有害健康的后果,包括肺损伤、低血压、肺塌陷、心脏骤停和死亡。
已收到3份关于该器械问题的投诉和1份伤害报告,目前还没有死亡报告。
受影响人群:
 使用高频输送Phasitron呼吸回路套件进行通气的人员。
使用高频输送Phasitron呼吸回路套件进行通气的人员。
 使用高频输送Phasitron呼吸回路套件为患者通气的医护人员。
使用高频输送Phasitron呼吸回路套件为患者通气的医护人员。
采取措施:
2023年12月12日,Percussionaire公司向所有受影响的客户(经销商和医护人员)发送了紧急现场安全通知。
通知要求经销商:
 完成并返回通知随附的经销商确认表
完成并返回通知随附的经销商确认表
 向最终客户发送包含预使用清单的紧急现场安全通知
向最终客户发送包含预使用清单的紧急现场安全通知
 优先退回库存中受影响批次的产品
优先退回库存中受影响批次的产品
 支持最终客户退回受影响的产品
支持最终客户退回受影响的产品
通知要求医护人员:
 完成并返回通知中随附的确认表
完成并返回通知中随附的确认表
 患者使用前填写预检查清单(包含在通知中)
患者使用前填写预检查清单(包含在通知中)
 优先退回所有受影响的产品
优先退回所有受影响的产品
(美国FDA网站)
澳大利亚TGA发布关于Smiths Medical公司因循环模式故障召回paraPAC plus呼吸机的警示信息
发布日期:2024年2月14日
召回级别:I类
召回产品:paraPAC plus 呼吸机
产品型号:310
产品代码:P310NAU
ARTG 334236(Smiths Medical Australasia Pty Ltd - 便携式气动呼吸机)
召回原因:当paraPAC plus呼吸机调节到“通气”操作模式时,会间歇性地提供持续正压气流,来代替预期的像人类一样的呼吸循环。
如果在循环模式下出现非循环并持续的正压气流则是一种故障,这使呼吸机不能按设定程序正常运行。
如果呼吸机持续提供正压气流,而不是预期的像人类一样的呼吸循环,可能会导致治疗延迟、没有通气、潮气量过大或压力过大。如果设备不能使患者在呼吸周期充分呼气,则可能导致缺氧。基于临床实际,这些情况可能会导致患者严重伤害或死亡。
召回措施:
目前正在研究该问题的解决方法,当能提供更多信息时,将与用户联系。
在此期间,Smiths Medical公司建议用户:
提高安全意识,遵循说明书中所有要求,包括用户手册中的警告和注意事项,包括但不限于以下内容:
 持续监测患者生命体征。
持续监测患者生命体征。
 单独监测脉搏血氧饱和度和呼末二氧化碳浓度。
单独监测脉搏血氧饱和度和呼末二氧化碳浓度。
 每次使用前必须完成所有使用前检查。
每次使用前必须完成所有使用前检查。
 在呼吸机失灵或发生故障时,必须提供替代通气方式,例如球囊面罩通气。
在呼吸机失灵或发生故障时,必须提供替代通气方式,例如球囊面罩通气。
(澳大利亚TGA网站)
美国FDA发布Medical ASD公司因早期软件版本问题召回Medfusion 4000型注射泵的警示信息
发布日期:2024年2月14日
召回级别:I级,是最严重的召回类型,使用这些产品可能造成严重伤害或者死亡。
产品名称:Medfusion注射泵
产品代码:FRN
产品型号:4000型注射泵
分销日期:2010年11月16日至2023年7月28日
在美国召回数量:50,743台
召回发起日期:2023年12月19日
产品用途:Medfusion 4000型注射泵专为精确控制输液速率而设计,使其适用于诸如血液、脂质、药物、抗生素和治疗液体等液体的管理。它适用于各种输送途径,包括动脉、硬膜外、静脉、鞘内、皮下和肠内。该泵支持连续、体积/时间、质量、体重、间歇和大剂量等输送模式。它适用于重症监护、麻醉、新生儿和儿科设置,以及任何需要使用注射泵进行临床监测或管理的医疗环境。
召回原因:由于注射泵早期软件版本相关的问题,Smiths Medical ASD公司正在召回Medfusion 4000型注射泵。与早期注射泵软件版本相关的问题可能会影响报警系统、泵、控制屏和泵的其他部分。如果设备中存在未检测到的问题,则设备可能会出现故障,导致治疗延迟或中断,或者设备可能无法按照编程设置提供治疗。
有1份与此问题相关的伤害报告,没有死亡报告。
受影响人群:
 使用Medfusion 4000型注射泵的医务人员。
使用Medfusion 4000型注射泵的医务人员。
 接受Medfusion 4000型注射泵治疗的患者人群,特别是易受伤害的患者,例如在新生儿重症监护病房(NICU)和心脏重症监护病房(CICU)的患者。
接受Medfusion 4000型注射泵治疗的患者人群,特别是易受伤害的患者,例如在新生儿重症监护病房(NICU)和心脏重症监护病房(CICU)的患者。
采取措施:2023年12月19日,Smiths Medical ASD公司发出了一封紧急医疗器械更正函。
信中要求客户:
 找到所有受影响的注射泵,并确保这些设备的所有用户立即关注此通知及解决措施。
找到所有受影响的注射泵,并确保这些设备的所有用户立即关注此通知及解决措施。
 确认所有注射泵都安装了最新的Medfusion软件。
确认所有注射泵都安装了最新的Medfusion软件。
 填写所提供的回复表格,并在收到本信函后10天内发送至smithmedical6114@sedgwick.com邮箱。
填写所提供的回复表格,并在收到本信函后10天内发送至smithmedical6114@sedgwick.com邮箱。
受影响设备的完整清单
之前使用以下软件版本的Medfusion注射泵可能存在以下问题:
 电机不运行触发高优先级报警(v1.0.0, v1.1.0, v1.1.1, v1.1.2)
电机不运行触发高优先级报警(v1.0.0, v1.1.0, v1.1.1, v1.1.2)
 重置给药剂量(v1.0.0, v1.1.0, v1.1.1, v1.1.2)
重置给药剂量(v1.0.0, v1.1.0, v1.1.1, v1.1.2)
 错误的紧急数据缺失报警
错误的紧急数据缺失报警
 中断给药或加载剂量(v1.0.0, v1.1.0, v1.1.1, v1.1.2)
中断给药或加载剂量(v1.0.0, v1.1.0, v1.1.1, v1.1.2)
 总剂量显示错误(v1.1.0, v1.1.1, v1.1.2)
总剂量显示错误(v1.1.0, v1.1.1, v1.1.2)
 给药/给药完成前的体积限制(v1.1.2)
给药/给药完成前的体积限制(v1.1.2)
 药物下限显示不正确(v1.6.0, v1.6.1)
药物下限显示不正确(v1.6.0, v1.6.1)
 电池耗尽警报(v1.6.5之前的所有版本)
电池耗尽警报(v1.6.5之前的所有版本)
 失去无线连接(v1.5.0, v1.5.1, v1.6.0, v1.6.1, v1.6.4)
失去无线连接(v1.5.0, v1.5.1, v1.6.0, v1.6.1, v1.6.4)
 PharmGuard服务器密码(v2.3, v2.4, v2.5)。
PharmGuard服务器密码(v2.3, v2.4, v2.5)。
(美国FDA网站)
美国FDA发布Philips公司因探头意外坠落召回BrightView成像系统的警示信息
发布日期:2024年2月15日
召回级别:I级,是最严重的召回类型,使用这些产品可能造成严重伤害或者死亡。
产品名称:BrightView,BrightView X,BrightView XCT
产品型号:
 BrightView:882480;453560279781,453560279791,453560279811,453560279801,2170-3000A,2170-3001A,2170-3002A,2170-3003A
BrightView:882480;453560279781,453560279791,453560279811,453560279801,2170-3000A,2170-3001A,2170-3002A,2170-3003A
 BrightView X:882478;453560824741,453560829261
BrightView X:882478;453560824741,453560829261
 BrightView XCT:882482;453560462131,453560749161
BrightView XCT:882482;453560462131,453560749161
生产日期:2007年9月至2013年6月
分销日期:2007年11月29日至2013年6月5日
在美国召回数量:553
召回发起日期:2023年12月15日
产品用途:Philips Brightview成像系统是一种单光子发射计算机断层扫描(SPECT)设备,作为医护人员的检查手段,拍摄和显示人体生物活动的图像。BrightView XCT型号产品结合了SPECT和计算机断层扫描(CT)成像两种功能。
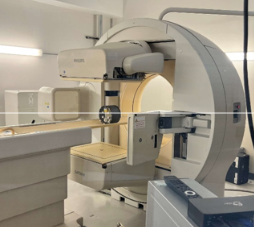
召回原因:Philips公司发现BrightView成像系统的探测器可能会因组件故障而意外坠落,决定召回BrightView、BrightView X和BrightView XCT型号的产品。意外坠落的探测器可能导致的人员伤害包括:颈部受伤、挫伤、创伤性脑损伤/脑震荡、死亡、挤压伤、骨折、撕裂伤、肌肉或韧带扭伤/拉伤等,同时导致BrightView系统运转中断。如果探测器位于设备的大开口上方(龙门架的中心),则可能会中断正常的系统运转。Philips公司收到使用上述设备的1份故障报告,没有伤亡报告。
受影响人群:
 使用Philips BrightView,BrightView X,BrightView XCT产品进行扫描的人群
使用Philips BrightView,BrightView X,BrightView XCT产品进行扫描的人群
 使用BrightView成像系统获得人体详细图片或检查结果的医护人员
使用BrightView成像系统获得人体详细图片或检查结果的医护人员
 负责维护Philips BrightView,BrightView X,BrightView XCT现场服务工程师
负责维护Philips BrightView,BrightView X,BrightView XCT现场服务工程师
采取措施:
2023年12月16日,Philips公司向所有受影响的客户发送了一封紧急医疗器械更正函。要求所有受影响的用户不要将患者的下肢直接放置在检测器下方,龙门架中心下方。如果探测器位于龙门架中心上方,且探测器支撑组件出现故障,则探测器将无法移动完成成像。
信函中还描述了两种可能的故障情形:
 情形1:探测器位于龙门架中心下方:如果患者的下肢直接位于下部探测器下方,且支撑部件发生故障,探测器可能会以不受控制的方式向下下降并接触患者。
情形1:探测器位于龙门架中心下方:如果患者的下肢直接位于下部探测器下方,且支撑部件发生故障,探测器可能会以不受控制的方式向下下降并接触患者。
 情形2:龙门架中心上方的探测器:如果支撑组件发生故障,探测器将保持原位,不会按照临床成像的预期移动,导致系统正常运转中断。可能需要对患者进行重新扫描或重新注射放射性药物。
情形2:龙门架中心上方的探测器:如果支撑组件发生故障,探测器将保持原位,不会按照临床成像的预期移动,导致系统正常运转中断。可能需要对患者进行重新扫描或重新注射放射性药物。
Philips公司宣布将联系客户,安排现场服务工程师(FSE)访问客户现场,并在必要时更正系统。
(美国FDA网站)



 加拿大Health Canada发布关于GE Healthcare公司召回Ic9-Rs超声探头的的警示信息
加拿大Health Canada发布关于GE Healthcare公司召回Ic9-Rs超声探头的的警示信息 美国FDA发布关于Cardinal Health公司因与注射泵不兼容召回一次性使用注射器的警示信息
美国FDA发布关于Cardinal Health公司因与注射泵不兼容召回一次性使用注射器的警示信息 美国FDA发布关于Percussionaire公司因超压召回高频输送Phasitron呼吸回路套件的警示信息
美国FDA发布关于Percussionaire公司因超压召回高频输送Phasitron呼吸回路套件的警示信息 澳大利亚TGA发布关于Smiths Medical公司因循环模式故障召回paraPAC plus呼吸机的警示信息
澳大利亚TGA发布关于Smiths Medical公司因循环模式故障召回paraPAC plus呼吸机的警示信息 美国FDA发布Medical ASD公司因早期软件版本问题召回Medfusion 4000型注射泵的警示信息
美国FDA发布Medical ASD公司因早期软件版本问题召回Medfusion 4000型注射泵的警示信息 美国FDA发布Philips公司因探头意外坠落召回BrightView成像系统的警示信息
美国FDA发布Philips公司因探头意外坠落召回BrightView成像系统的警示信息