FDA调查接受靶向BCMA或CD19的自体嵌合抗原受体(CAR)-T细胞免疫治疗后发生T细胞恶性肿瘤的严重风险
美国食品药品管理局(FDA)发布消息,已收到接受靶向BCMA或CD19的自体CAR-T细胞免疫治疗后患者出现T细胞恶性肿瘤(包括嵌合抗原受体CAR阳性淋巴瘤)的报告。这些报告来自临床试验和/或上市后不良事件(AE)。
FDA 已确定,T细胞恶性肿瘤的风险适用于目前批准的所有经基因修饰的靶向BCMA和靶向CD19的自体CAR-T细胞免疫疗法。接受过多种同类产品治疗的患者发生了 T 细胞恶性肿瘤。目前获批的此类产品(按商品名称字母顺序排列)包括:
 Abecma (idecabtagene vicleucel)
Abecma (idecabtagene vicleucel)
 Breyanzi (lisocabtagene maraleucel)
Breyanzi (lisocabtagene maraleucel)
 Carvykti (ciltacabtagene autoleucel)
Carvykti (ciltacabtagene autoleucel)
 Kymriah (tisagenlecleucel)
Kymriah (tisagenlecleucel)
 Tecartus (brexucabtagene autoleucel)
Tecartus (brexucabtagene autoleucel)
 Yescarta (axicabtagene ciloleucel)
Yescarta (axicabtagene ciloleucel)
虽然此类产品用于获批适应症的总体获益仍然大于潜在风险,但FDA正在调查导致住院和死亡等严重后果的T细胞恶性肿瘤风险,并评估是否采取监管措施。
与所有使用整合性载体(慢病毒或逆转录病毒载体)的基因治疗产品一样,继发性恶性肿瘤潜在风险是已获批准的靶向BCMA和靶向CD19的基因修饰自体CAR-T细胞免疫疗法的美国处方信息 (USPIs) 中的警告信息。根据《联邦食品、药品和化妆品法案》(Federal Food, Drug, and Cosmetic Act, FDCA)第 505(o)条的规定,这些产品最初批准上市时要求开展上市后研究(PMRs),包括一个为期 15 年的安全性长期随访观察研究,以评估长期安全性和治疗后发生继发性恶性肿瘤的风险。
接受这些产品治疗的患者和临床试验参与者应终身监测新发恶性肿瘤。如果在使用这些产品治疗后出现新发恶性肿瘤,请联系生产商报告该事件,并获取有关患者采集样本以检测是否存在嵌合抗原受体 (CAR) 转基因的指导。
(美国食品药品管理局FDA网站)
原文链接:
https://www.fda.gov/vaccines-blood-biologics/safety-availability-biologics/fda-investigating-serious-risk-t-cell-malignancy-following-bcma-directed-or-cd19-directed-autologous
加拿大修订拉罗替尼说明书增加肝毒性等风险
加拿大最近对拉罗替尼(larotrectinib,商品名Vitrakvi)产品专论进行修订,在用法用量、不良反应、注意事项和药物相互作用部分已更新“肝毒性和涉及中度细胞色素P450(CYP)3A4诱导剂的药物相互作用”风险信息。
给医务人员的信息:
1.在接受拉罗替尼治疗的成年患者中,有丙氨酸氨基转移酶(ALT)和/或天冬氨酸转氨酶(AST)2、3或4级严重程度升高以及胆红素升高≥2 x ULN(正常上限)的肝毒性病例报告。
2.首次给药前,应对转氨酶水平在内的肝功能进行基础评估。治疗期间监测肝功能包括ALT、AST、碱性磷酸酶(ALP)和胆红素检查(具体内容请参阅产品专论注意事项)。
3.在肝转氨酶升高的患者中,根据其严重程度,采取停用、调整剂量或永久停用拉罗替尼的措施(具体内容请参阅产品专论用法用量)。
4.拉罗替尼是CYP3A的底物。拉罗替尼与中度(或强效)CYP3A4诱导剂联合给药可能会降低拉罗替尼的血浆浓度。
参考:拜耳公司的拉罗替尼产品专论详见链接https://pdf.hres.ca/dpd_pm/00072102.PDF。
(加拿大卫生部网站)
原文链接:
https://www.canada.ca/en/health-canada/services/drugs-health-products/medeffect-canada/health-product-infowatch/november-2023.html#a3.2.1
欧洲药品管理局修改洛莫司汀的血小板减少症及过量用药信息
欧洲药品管理局的药物警戒风险评估委员会(PRAC)在对洛莫司汀(lomustine)的定期安全更新报告评估后,认为:基于文献和自发报告中的现有数据,洛莫司汀与过量用药、与血小板减少症之间的因果关系存在合理可能性,含有洛莫司汀的产品信息应相应修改。欧盟人用药品互认和分散程序协调小组(CMDh)审查后同意以上结论和建议。欧盟各成员国于2024年1月25日执行以上措施。
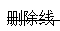 批准的产品信息修订内容如下(新添文本下划线,粗体,删除文本
批准的产品信息修订内容如下(新添文本下划线,粗体,删除文本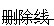 ):
): 药品说明书
第4.4节
…
必须严格指导患者不要服用比医生建议剂量更高的剂量,并应告知患者该药是作为单次口服剂量(或三天内的分剂量)服用的,至少6周内不要重复服药(见第4.2节)。
第4.8节
不良反应 血小板减少症的频率应改为非常常见。
包装标签
第2节
警告和注意事项
…
应完全按照医生的处方剂量服用本品,至少在6周内不要重复服用。
第3节
如果超剂量服用,应立即就医。有报道意外过量服用导致死亡的病例。服药过量可能表现为腹痛、腹泻、返流、食欲不振、嗜睡、头晕、咳嗽或呼吸急促、不明原因的瘀伤或出血或容易感染。
第4节
以下不良反应发生频率应从“未知”转移到“非常常见”:血小板水平低,可导致出血和瘀伤。
(EMA网站)
原文链接:
https://www.ema.europa.eu/en/documents/psusa/lomustine-cmdh-scientific-conclusions-and-grounds-variation-amendments-productinformation-and-timetable-implementation-psusa-00001902-202301_en.pdf
欧洲药品管理局修改依托泊苷的机会性感染和使用内联过滤器时增加过敏反应的风险信息
欧洲药品管理局的药物警戒风险评估委员会(PRAC)近期在对依托泊苷(etoposide)的定期安全更新报告评估后,认为:基于临床试验、文献和自发报告中的现有数据,依托泊苷与机会性感染(如吉氏肺孢子虫肺炎)之间的因果关系存在合理可能性;基于文献中关于使用内联过滤器(In-line filters)给药时超敏反应风险增加的现有数据,使用内联过滤器给予依托泊苷(而非磷酸依托泊苷)与这种风险增加之间存在因果关系的合理可能性;含有依托泊苷的产品信息应做出修改。欧盟人用药品互认和分散程序协调小组(CMDh)审查后同意以上结论和建议。欧盟各成员国于2024年1月25日执行以上措施。
批准的产品信息修订内容如下(新添文本下划线,粗体,删除文本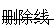 ):
):
药品说明书
第4.4节
警告应修改如下(仅适用于注射/输注产品,不适用于含磷酸依托泊苷的产品):
超敏反应
医生应注意可能发生的过敏反应,表现为寒冷、发热、心动过速、支气管痉挛、呼吸困难和低血压,有可能致命。有症状时应立即停止使用药品,随后在医嘱下给予升压药、皮质类固醇、抗组胺药或扩容剂。当使用内联过滤器进行依托泊苷给药,观察到输液相关超敏反应的风险增加时,则不应再使用内联过滤器。
第4.8节
所有含有依托泊苷或磷酸依托泊苷的产品应修改以下SOC(感染和侵袭)项下不良反应:
感染*
*包括机会性感染,如吉氏肺孢子虫肺炎
包装标签
第2节
可能的副作用
感染(包括免疫系统减弱患者的感染,例如称为吉氏肺孢子虫肺炎的肺部感染)
(EMA网站)
原文链接:
https://www.ema.europa.eu/en/documents/psusa/etoposide-cmdh-scientific-conclusions-and-grounds-variation-amendments-product-information-and-timetable-implementation-psusa-00001333-202302_en.pdf


